Gene Targeting and Transgenic Models Platform
OVERVIEW
IBG Gene Targeting and Transgenic Models Platform has recently established its research laboratories and state of the art technological infrastructure. Our platform will provide services and collaborative support to internal and external researchers working in basic and translational biomedical sciences, whose work involve or require the generation of genetically-engineered mouse models.
In addition to the services we offer below, our lab is interested in investigating rare genetic disorders utilizing transgenic mouse models.
COLLABORATIVE SUPPORT AND SERVICES
![]()
Generation of knockout and knockin mouse models:
Our research platform invites researchers to collaborate with us for the generation of knockout and knockin mouse models for their favourite genes. We offer collaborations starting from initial planning and vector design stage to the final establishment of a genetically modified mouse colony.
We have, quite recently, created our first knockin mouse model for a new rare genetic disorder (see News tab). We are currently developing several transgenic disease models. We are ready to collaborate with interested partners who need to generate novel genetically modified mouse models.
Mouse embryo cryopreservation and rederivation:
The technological infrastructure for embryo cryopreservation and rederivation has been recently completed. After establishment of the relevant protocols, we will provide services to internal and external users who wish to store their transgenic mouse strains long term.
SERVICES - Currently provided
Services we currently provide are listed below. Please contact us via e-mail for further details, pricing and conditions of the services.
1. Knockout (indel)
2. Knockout (deletion)
3. Knockin (point mutations)
4. Knockin (epitope tags)
5. ESC gene targeting
6. Sperm cryopreservation
7. Embryo transfer and mouse strain rederivation
SERVICES - In development
The services listed below are within our technical expertise, although we do not routinely apply these protocols.
1. Embryo cryopreservation
2. IVF (in vitro fertilization)
3. ESC microinjection
4. PNI (pronuclear injection)
Personnel

Gene Targeting and Transgenic Models Platform
Research Group Leader
Kasım DİRİL
kasim.diril@ibg.edu.tr
+90 232 299 41 00
(5091)
+9
0 232 299 41 59

Kerem ESMEN
R&D Personnel
kerem.esmen@ibg.edu.tr

Seval KILIÇ
PhD Student
seval.kilic@ibg.edu.tr

Aslı BABAYAKALI
MSc Student
asli.babayakali@ibg.edu.tr

Nazlıcan KESMİK
Undergraduate Student
nazlican.kesmik@std.ibg.edu.tr

Emre KALAY
MSc Student
emre.kalay@std.ibg.edu.tr

Keziban KORKMAZ BAYRAM
Researcher
None
Former Personnel

Mehmet ERGÜVEN
Visiting Researcher
mehmet.erguven@msfr.ibg.edu.tr
Mehmet ERGÜVEN
MSc Student
mehmet.erguven@msfr.ibg.edu.tr

Liubovi SOPCO
Undergraduate Student
liubovi.sopco@msfr.ibg.edu.tr

Gizem ÖZTÜRK
MSc Student
gizem.ozturk@ibg.edu.tr
Liubovi SOPCO
Undergraduate Student
liubovi.sopco@msfr.ibg.edu.tr

Gülin KÖKLÜ
Undergraduate Student
gulin.koklu@ibg.edu.tr

Ferzane VALİOĞLU
Post-Doc Researcher
ferzane.valioglu@ibg.edu.tr

Pelin Dide BEŞER
Undergraduate Student
pelindide.beser@ibg.edu.tr
Gizem ÖZTÜRK
PhD Student
gizem.ozturk@ibg.edu.tr

Cengizhan BİLGİN
Undergraduate Student
cengizhan.bilgin@ibg.edu.tr

Abdullah ERİM
Undergraduate Student
abdullah.erim@ibg.edu.tr
Liubovi SOPCO
MSc Student
liubovi.sopco@msfr.ibg.edu.tr

Tansu Bilge KÖSE
PhD Student
None

Ayşe Miray OTO
Undergraduate Student
miray.oto@ibg.edu.tr

Ece Şule ZANGUR
MSc Student
ece.zangur@ibg.edu.tr
News
NanoBio4Can postdoctoral programme

We are looking for motivated postdoctoral candidates who will establish a novel humanized mouse model for liver cancer research.
https://nanobio4can.net/about-us/programme-description
About the project:
Hepatocellular carcinoma manifests high genetic heterogeneity between patients as well as cancer cells isolated from the very same tumour. The project we offer in the NanoBio4Can programme aims to develop an in vivo personalized medicine platform, very much similar to “avatar mice”, for testing drugs and modelling disease using tumor cells derived from patients. An FRG triple knockout transgenic mouse line will be developed in house and utilized for implantation and homing of patient tumors into the mouse liver. Knockout lines for FAH, Rag2 and Il2rg have already been created. Within the scope of the project, additional genetic modifications as well as allele combinations may be explored that allow efficient humanization of the liver and immune system, with the final aim of efficiently populating the mouse liver with patient derived tumour cells.
FRG triple-knockout: A mouse model whose liver can be humanized
We have recently gained support from TUSEB for a project which aims to develop a sophisticated mouse model. These mice allow engraftment of human hepatocytes into their livers.
In our project; knockout alleles will be created by introducing mutations in FAH, RAG2, IL2RG genes and subsequently all KO alleles will be combined in a single mouse line (FRG-TKO).
FLAG- tagged knockin mice
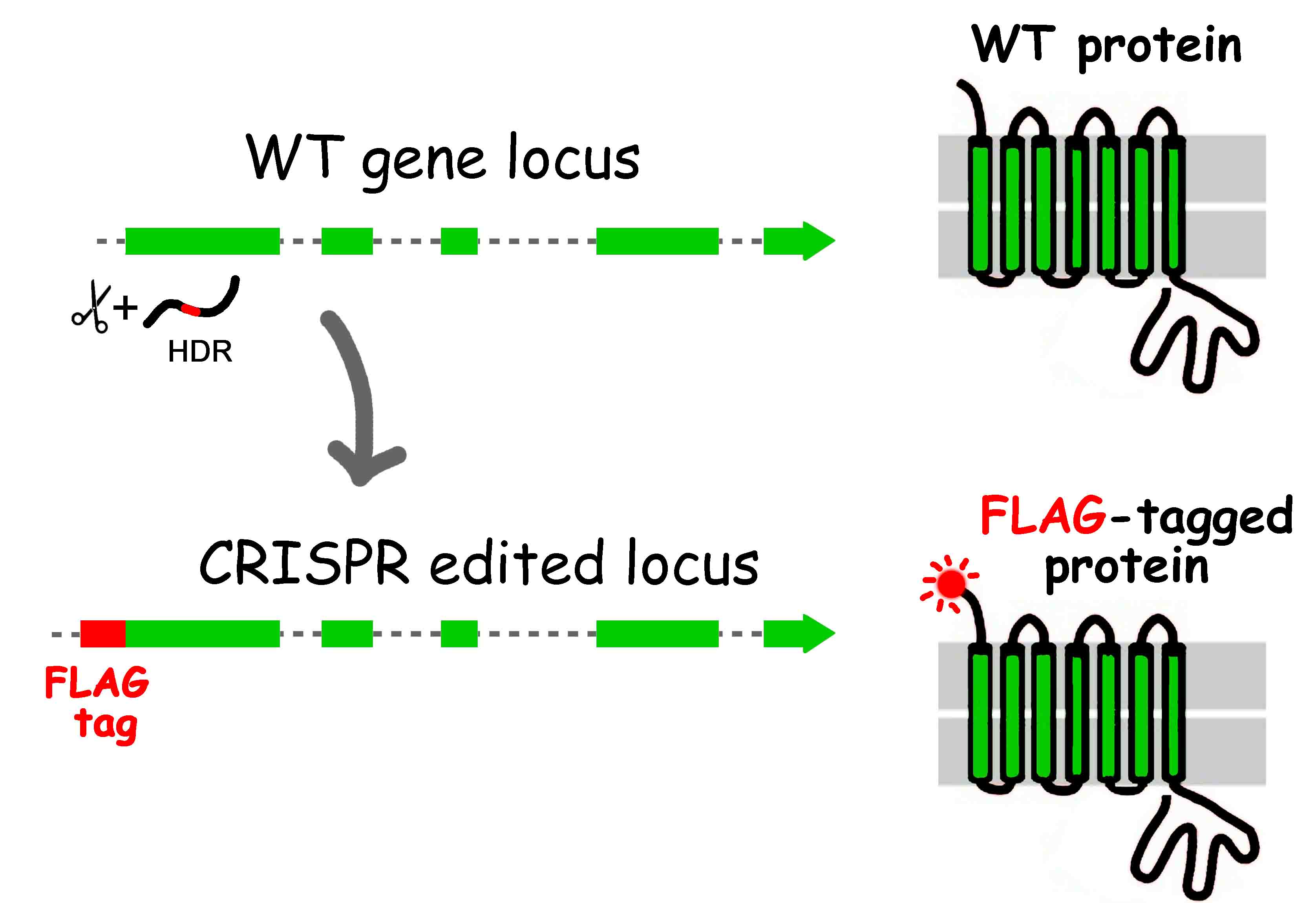
We have generated a transgenic line that expresses an N-terminal FLAG-epitop tagged transmembrane protein. A 24-nucleotide long DNA sequence, encoding the DYKDDDDK peptide tag, was introduced to the first coding exon of the gene by CRISPR HDR-mediated knockin.
These mice will produce the FLAG-tagged version of the protein which will be expressed from the endogenous gene locus. Therefore, it will be possible to determine the tissue specific/developmental expression and subcellular distribution of the endogenous gene/protein using the very specific anti-FLAG antibodies.
27 December 2022
IFNAR1 KO mice
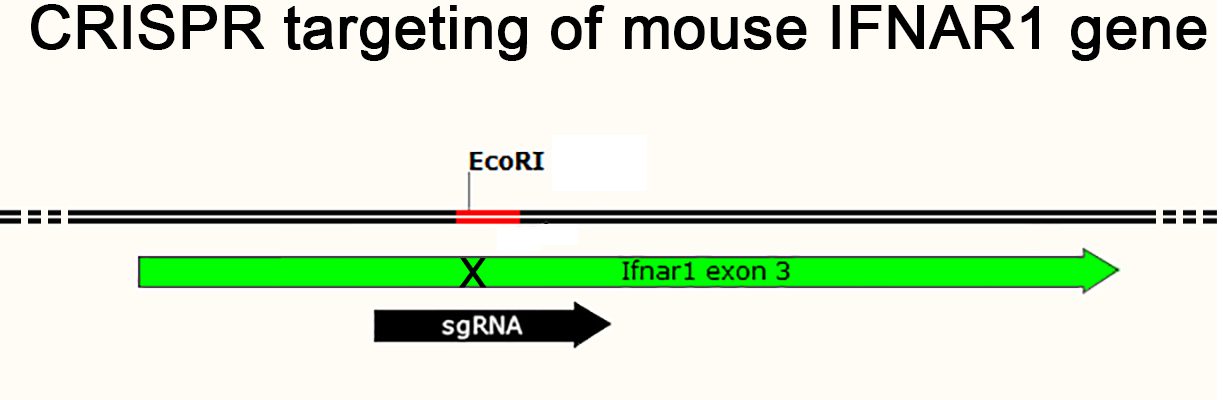
IBG Transgenic Platform has developed the IFNAR1 knockout mouse model. The IFNAR1 gene codes for the type-I interferon-α/β receptor, which activates JAK-STAT signaling pathways upon binding type-I IFN. JAK-STAT signaling is essential for processes including growth regulation, survival, differentiation, pathogen resistance and antiviral immune responses. IFNAR1 knockout mice do not display type-I IFN receptor function which causes reduced immune response and an increased susceptibility to viral infection. IFNAR1 knockout mice are widely used in research on antiviral immune responses, and interferon stimulation and JAK-STAT signaling.
Expanding our transgenic models repertoire

Recently, we have added two knockin and one knockout mouse models to our platform's repertoire. The total number of home-made transgenic models is now five.
One of the knockin strains we engineered, will be used to study a newly defined neurodevelopmental rare genetic disorder.
The other mouse models are for the investigation of gene functions in vivo.
At this stage, we can confidently offer knockin or knockout models as a service to academia.
Our first rare genetic disease model was created
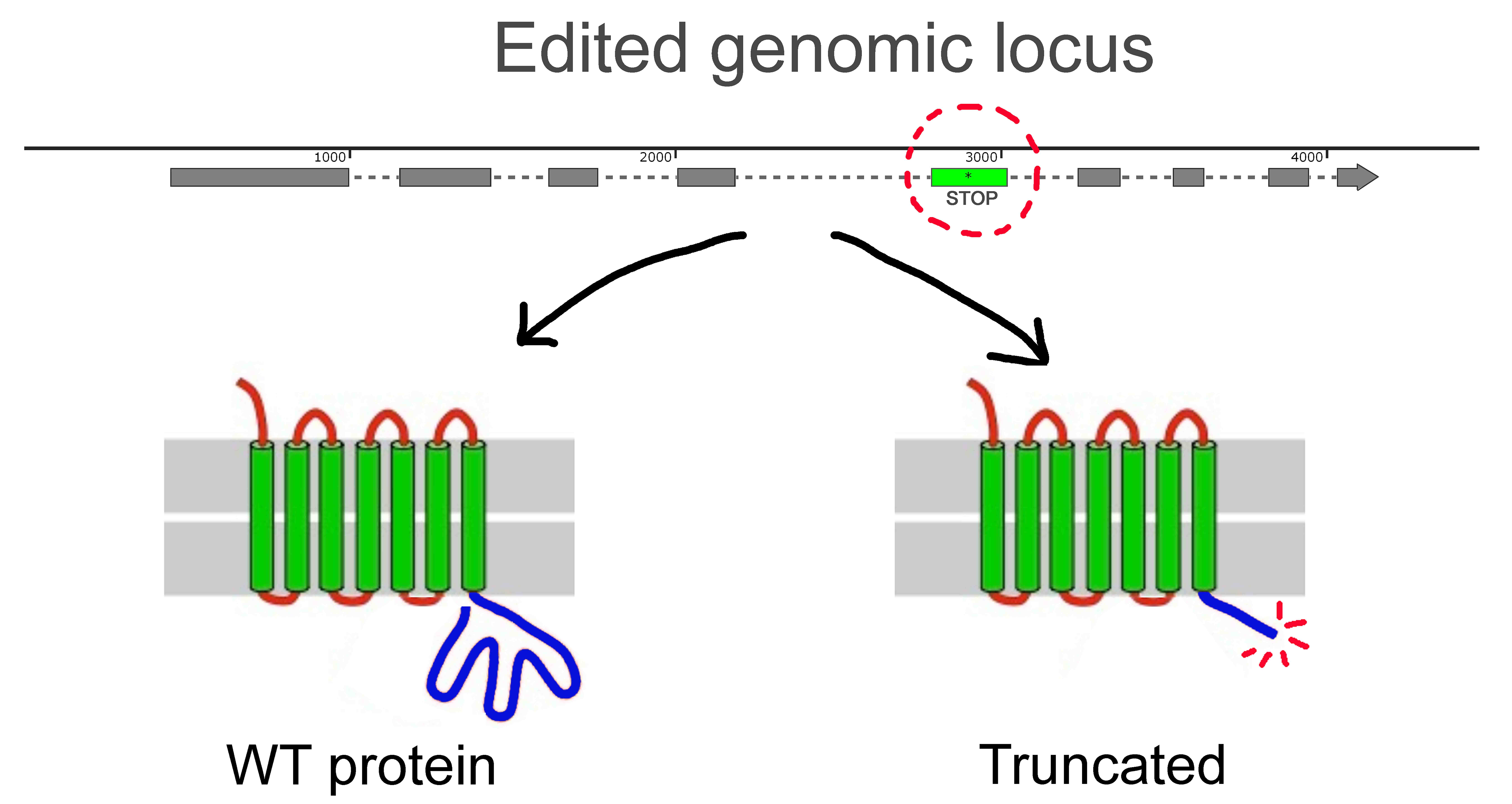
We have recently created our first transgenic model for a novel rare genetic disease. The mutation in the patients causes truncation of the cytoplasmic tail domain of a multi-pass transmembrane protein. To mimic the mutation in mice, we have engineered the mouse genomic locus and introduced a STOP codon into the relevant coding exon. This results in truncation of the protein product- a transmembrane protein with a shorter cytosolic domain. These mice will serve as an in vivo model for the characterization of the disease.
12 December 2021
Albino C57BL/6J transgenic mice
Our platform has recently created a genetically modified (transgenic) mouse model for the first time in our laboratory. We have edited the Tyr gene in C57BL/6J mice by CRISPR genome engineering technology. The knockin mutation we introduced, causes loss of function in the tyrosinase enzyme, resulting in albino skin color.
C57BL/6J is the most widely used mouse strain in biomedical research. Albino C57BL/6J mice are the preferred model for bioluminescence imaging studies due to their lack of skin pigment melanin. Additionally, their embryos are commonly used for microinjection of ES cells derived from BL6 strains. This newly created strain will be a valuable resource for IBG Vivarium and our transgenic platform.
We are currently in the process of creating several knockin mouse lines to model rare genetic disease in vivo.
Our ongoing work is funded by the TUBITAK 1005 program.
28 September 2021
Selected Publications
Diril MK, Erguven M. Genetic dissection of the Mastl-Arpp19/ENSAPP2A-B55δ pathway in mammalian cells. Turkish Journal of Biochemistry. 2023 April ; 48 (2) : 1-13. doi:10.1515/tjb-2022-0191. Download
Abu Alhaija AA, Lone IN, Ozkuru Sekeroglu E, Batur T, Angelov D, Dimitrov S, Hamiche A, Firat Karalar EN, Ercan ME, Yagci T, Alotaibi H, Diril MK. Development of a mouse embryonic stem cell model for investigating the functions of the linker histone H1-4.. FEBS open bio. 2023 December . doi:10.1002/2211-5463.13750. Download
Erguven M, Kilic S, Karaca E, Diril MK.. Genetic complementation screening and molecular docking give new insight on phosphorylation-dependent Mastl kinase activation. Journal of Biomolecular Structure and Dynamics. 2022 October . doi:10.1080/07391102.2022.2131627. Download
Erguven M, Karakulak T, Diril MK, Karaca E. How Far Are We from the Rapid Prediction of Drug Resistance Arising Due to Kinase Mutations?. ACS omega. 2021 January ; 6 (2) : 1254-1265. doi:10.1021/acsomega.0c04672. Download
Szmyd R, Niska-Blakie J, Diril MK, Renck Nunes P, Tzelepis K, Lacroix A, van Hul N, Deng LW, Matos J, Dreesen O, Bisteau X, Kaldis P.. Premature activation of Cdk1 leads to mitotic events in S phase and embryonic lethality. Oncogene. 2019 February ; 38 (7) : 998-1018. doi:10.1038/s41388-018-0464-0. Download
Total : 5
Selected Proceedings
Seval Kılıç, Semra Hız, Kerem Esmen, Gökhan Karakülah, Mehmet Öztürk, Kasım Diril 2023, 'Generation and Phenotypic Characterization of a Transgenic Mouse Model for a Neurodevelopmental Rare Genetic Disorder', 47th FEBS Congress, Fransa, July 8, 2023 . Download
Total : 1
Projects
The Scientific and Technological Research Council of Turkey - TUBITAK - RD : Analysis of mitotic functions of ENSA and Arpp19 proteins, Finished
DEÜ BAP - Dokuz Eylul University Scientific Research Projects Coordination Unit - RD : Mastl kinazın mitotik fonksiyonlarının kimyasal genetik yöntemlerle analizi, Finished
The Scientific and Technological Research Council of Turkey - TUBITAK - RD : Transgenik fare modellerinin yerel üretimi için yöntem geliştirilmesi, Ongoing
The Scientific and Technological Research Council of Turkey - TUBITAK - RD : Generation and phenotypic characterization of a transgenic mouse model for a neurodevelopmental rare genetic disorder, Ongoing
Health Institutes of Turkey- TUSEB - RD : Generation and basic phenotypic characterization of a mouse model of a rare neurogenetic disease observed in Turkish population: TRAPPC4 mutation, Funded, Not Started
Health Institutes of Turkey- TUSEB - RD : Development of FRG knockout humanized liver mouse model, Ongoing
Open Positions
NanoBio4Can programme, postdoctoral candidates:
Please see the news tab on the left side panel!
Postdoctoral researcher
We are looking for a postdoctoral researcher to join our research team working on mouse models of rare genetic disorders.
The candidate is expected to apply to the TÜBİTAK BİDEB 2218 program for financial support.
The project proposal should involve creation of a transgenic mouse model for a novel or known rare genetic disorder with subsequent phenotyping analyses. Help and guidance will be provided for selecting a suitable project topic and writing of the proposal.
Candidates should i) have a very good command of English, ii) able and willing to work with mouse models, and iii) be experienced in basic molecular biology techniques (PCR, cloning, restriction analysis etc.).
Please see the details in the link provided below (in Turkish).
Graduate Students: We are looking for graduate students (MSc/PhD) with a very good level of English. Please see above for other criteria.
Veterinary Technician: For colony management. Should have a certificate for experimental animal use and experience with rodents.
Awards
- Young Investigator (GEBİP) Awards by Turkish Academy of Sciences (TÜBA), 2017
- Young Scientist (BAGEP) Award by Science Academy, 2016
- 2232 Yurda Dönüş Araştırmacı Dolaşım Programı by TUBITAK, 2015
Contact
Gene Targeting and Transgenic Models Platform
Research Group Leader
Kasım DİRİL
kasim.diril@ibg.edu.tr
+90 232 299 41 00
(5091)
+9
0 232 299 41 59